

Glykogenstruktur, Synthese, Abbau, Funktionen
- 1254
- 342
- Lewis Holzner
Er Glykogen Es ist das Lagerkohlenhydrat der meisten Säugetiere. Kohlenhydrate werden üblicherweise als Zucker bezeichnet und diese werden gemäß der Anzahl der durch Hydrolyse verursachten Abfälle (Monosaccharide, Disaccharide, Oligosaccharide und Polysaccharide) klassifiziert).
Monosaccharide sind die einfachsten Kohlenhydrate, die gemäß der Anzahl der in ihrer Struktur enthaltenen Kohlenhäuser klassifiziert sind. Dann gibt es die Triosas (3C), Tetrosas (4C), Pentosas (5C), Hexosous (6C), Heptosase (7C) und Oktosas (8c).
Chemische Glykogenstruktur zeigt Glykosidbindungen (Quelle: Glykogen.SVG: Neurotoger -Derivatarbeit: Marek M [Public Domain] über Wikimedia Commons) Nach dem Vorhandensein der Aldehydgruppe oder der Cetona -Gruppe werden diese Monosaccharide ebenfalls als Aldies oder Ketosas klassifiziert.
Disaccharide führen zu Hydrolyse, zwei einfache Monosaccharide, während Oligosaccharide 2 bis 10 Einheiten Monosaccharide und Polysaccharide produzieren, die mehr als 10 Monosaccharide produzieren.
Der Glykogen ist aus biochemischer Sicht ein Polysaccharid, das aus verzweigten Ketten einer Sechs -Kohlenstoff -Aldose besteht, dh einer Hexose, die als Glukose bekannt ist. Grafisch kann es als Glukosebaum als Glykogen dargestellt werden. Dies wird auch als Tierstärke bezeichnet.
Glukose in Pflanzen wird als Stärke und bei Tieren als Glykogen gespeichert, das hauptsächlich im Leber- und Muskelgewebe gespeichert ist.
In der Leber kann Glykogen 10% seiner Masse und 1% der Muskelmasse festlegen. Wie in einem 70 kg -Mann wiegt die Leber etwa 1800 g und die Muskeln etwa 35 kg, ist die Gesamtmenge des Muskelglykogens viel größer als die Leber.
[TOC]
Struktur
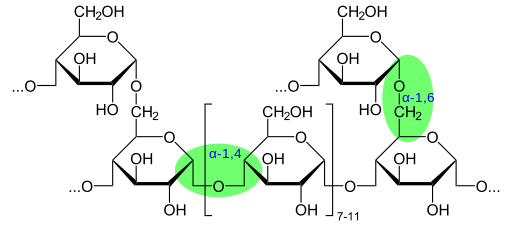
Das Molekulargewicht von Glykogen kann 108 g/mol erreichen, äquivalent zu 6 × 105 Glukosemolekülen. Glykogen besteht aus mehreren verzweigten α-D-Glycose-Ketten. Glucose (C6H12O6) ist eine Aldohexosa, die linear oder zyklisch dargestellt werden kann.
Der Glykogen hat eine sehr verzweigte und kompakte Struktur mit Ketten von 12 bis 14 Glucoseabfällen in Form von α-D-Glucose, die mit α- (1 → 4) glucosidischen Bindungen verbunden sind. Die Kettenrohrungen werden durch α- (1 → 6) glucosidische Glieder gebildet.
Glykogen liefert wie die in der Ernährung aufgenommene Stärke die meisten Kohlenhydrate, die der Körper benötigt. Im Darm werden diese Polysaccharide durch Hydrolyse abgebaut und dann hauptsächlich als Glukose in Richtung des Kreislaufs absorbiert.
Drei Enzyme: ß-Amylase, α-Amylase und Amylo-α- (1 → 6) -Glucosidase sind für den Darmabbau von Glykogen und Stärke verantwortlich.
Die α-Amylase hydrolysiert zufällig die α-Bindungen (1 → 4) der Seitenketten von Glykogen und Stärke und erhält daher den Namen der Endoglysidase. Die ß-Amyla ist eine Exoglicosidase, die ß-maltose dímeros freisetzt.
Angesicht.
Das Grenze Dextrin wird schließlich an den Zweigstellen hydrolysiert. Die durch diesen Defleat freigesetzten Ketten werden nach ß-Amylase und α-Amylase abgebaut.
Wenn der aufgenommene Glykogen als Glukose eintritt.
Kann Ihnen dienen: Purines: Eigenschaften, Struktur, FunktionenSynthese
Die Glykogensynthese wird als Glykogenese bezeichnet und findet insbesondere in Muskel und Leber statt. Die Glukose, die mit der Ernährung in den Organismus gelangt.
Glukochinase Phosphoryyl zu Glucose in Kohlenstoff 6. Das ATP liefert Phosphor und Energie für diese Reaktion. Infolgedessen wird Glukose 6-phosphat gebildet und ein ADP freigesetzt. Dann wird 6-phosphat-Glucose durch die Wirkung einer Phosphoglucomutase Glukose 1-phosphat.
Die 1-phosphat-Glucose wird für die Glykogensynthese aktiviert, was die Teilnahme eines Satzes von drei anderen Enzymen impliziert.
Glucose-1-phosphat zusammen mit dem Trifosphat-Uridin (UTP, einem Nucleosid von Uridintriphosphat) und durch Wirkung des UDP-Glycose-Pyrophosphorylase bilden das Diphosphat-Glucose-Uridinkomplex (UDP GLC) (UDP GLC). Dabei ist ein Pyrophosphation hydrolysiert.
Dann bildet das synthetierte Glykogenenzym eine glucosidische Bindung zwischen dem C1 des UDP -GLC -Komplexes und dem C4 eines Glykogenglukosetuchs, und das UDP -UDP wird aus dem UDP -aktivierten Glucosekomplex freigesetzt. Damit diese Reaktion auftritt, muss ein vorhandenes Glykogenmolekül als "primärer Glykogen" namens "Primärglykogen" vorhanden sein.
Das ursprüngliche Glykogen wird auf einem Priming -Protein Glykogenin synthetisiert, das 37 kDa und Glysila in einem Tyrosinrest unter Verwendung des UDP -GLC -Komplexes aufweist. Von dort aus sind sie mit 1 → 4-Glieder mit α-D-Glucoseabfällen verknüpft, und eine kleine Kette wird gebildet, auf die der Synthesaseglykogen wirkt.
Sobald die Anfangsketten mindestens 11 Glukosereste miteinander verbindet, überträgt das Verzweigungs- oder Amilenzym (1,4 → 1,6) -Glicosyltransferase ein Kettenstück von 6 oder 7 Glukoseabfällen an die angrenzende Kette in Position 1 → 6, die einen Zweig festlegen Punkt. Das so gebaute Glykogenmolekül wächst durch Zugabe von Glukoseeinheiten mit glycosidischen Verbindungen 1 → 4 und weitere Auswirkungen.
Degradierung
Glykogenabbau wird als Glucogenolyse bezeichnet und entspricht nicht dem umgekehrten Pfad seiner Synthese. Die Geschwindigkeit dieser Route ist durch die Geschwindigkeit der Reaktion begrenzt, die durch den Phosphorylaseglykogen katalysiert wurde.
Phosphorllaseglykogen ist für die Aufteilung (Phosphorolyse) von Links 1 → 4 aus Glykogenketten und Freisetzung von Glukose-1-Phosphat verantwortlich. Die enzymatische Wirkung beginnt an den Enden der äußersten Ketten und werden nacheinander entfernt, bis 4 Glukosereste auf jeder Seite der Auswirkungen bleiben.
Dann verlässt ein anderes Enzym, das α- (1 → 4) → α- (1 → 4) Glucano-Transferas, den Zweigpunkt aus. Dies ermöglicht die Amilo- (1 → 6) -Glucosidase (Unrampic Enzym) Hydrolys. Die kombinierte Wirkung dieser Enzyme endet vollständig auf Glykogen auf.
Da die anfängliche Reaktion der Phosphomutase reversibel ist, kann 6-phosphat-Glucose aus Glukoseresten 1-phosphat aus Glykogen gebildet werden. In Leber und Niere, aber nicht im Muskel, gibt es ein Enzym, Glucose-6-Phosphatase, das sich zu 6-phosphat-Glukose versammeln und in freie Glukose verwandelt.
Kann Ihnen dienen: FotolyseDefosphorylierte Glukose kann sich auf Blut ausbreiten, und so spiegelt sich die Glykogenolyse der Leber in einem Anstieg der Blutzuckerwerte (Glykämie) wider (Glykämie).
Regulierung der Synthese und Abbau
Der Synthese
Dieser Prozess wird auf zwei grundlegenden Enzymen ausgeübt: Synthesaseglykogen- und Phosphorylaseglykogen, so dass der andere, wenn einer von ihnen aktiviert ist, in seinem inaktiven Zustand ist. Diese Verordnung verhindert, dass gegen Synthese und Verschlechterung gleichzeitig auftreten.
Die aktive Form und die inaktive Form beider Enzyme sind sehr unterschiedlich, und die Interkonversion der aktiven und inaktiven Formen von Phosphorylase und synthetischer Glykogen unterliegt einer strengen hormonellen Kontrolle.
Adrenalin ist ein Hormon, das aus dem Nebennierenmark freigesetzt wird, und Glucagon ist ein weiteres, der im endokrinen Teil der Bauchspeicheldrüse auftritt. Endokrine Bauchspeicheldrüse produziert Insulin und Glucagon. Langerhans Insels α sind diejenigen, die den Glucagon synthetisieren.
Adrenalin und Glucagon sind zwei Hormone, die freigesetzt werden, wenn Energie als Reaktion auf die Abnahme des Blutzuckerspiegels benötigt wird. Diese Hormone stimulieren die Aktivierung von Phosphorylaseglykogen und hemmen Synthesaseglykogen, wodurch die Glykogenolyse stimuliert und die Glykogenese hemmt.
Während Adrenalin seine Wirkung auf Muskeln und Leber ausübt, wirkt Glucagon nur auf die Leber. Diese Hormone werden mit spezifischen membranalen Rezeptoren in der weißen Zelle verbunden, die Cyclasa Adenilat aktiviert.
Die Aktivierung des Cyclase -Adenylats beginnt einen enzymatischen Wasserfall, der einerseits eine AMPC -abhängige Proteinquinase aktiviert, die zu synthetischer Glykogen inaktiv ist und die Glykogenphosphorylase durch Phosphorylierung aktiviert (direkt bzw. indirekt).
Der Skelettmuskel verfügt.
Der Verschlechterung
Die oben beschriebenen enzymatischen Wasserfälle erhöhen den Glukosespiegel, und wenn sie ein bestimmtes Niveau erreichen.
Die Glykogenese wird durch Aktivierung der Phosphatase -Phosphorylase aktiviert, einem Enzym, das die Glykogensynthese durch mehrere Mechanismen reguliert.
Insulin fördert den Eintritt von Glukose in Muskelzellen und erhöht die 6-phosphat-Glukosespiegel, was die Defosphorylierung und Aktivierung von Synthesaseglykogen stimuliert. Somit beginnt die Synthese und der Glykogenabbau wird inhibiert.
Funktionen
Muskelglykogen stellt eine Energiereserve für den Muskeln dar, die es Muskeln ermöglicht, seine Funktionen zu erfüllen. Als Glukosequelle wird Muskelglykogen während des Trainings verwendet. Diese Reservierungen nehmen mit körperlichem Training zu.
In der Leber bildet Glykogen auch eine wichtige Reservequelle für sowohl die Funktionen des Organs als auch für den Beitrag von Glucose zum Rest des Körpers.
Diese Funktion des Leberglykogens ist auf die Tatsache zurückzuführen. Freie Glukose kann im Gegensatz zu phosphorylierten Glukose durch die Hepatozytenmembran (Leberzellen) verteilt werden.
Es kann Ihnen dienen: Sporulation: in Pflanzen, in Pilzen und in BakterienAuf diese Weise kann die Leber Glukose für den Kreislauf liefern und den stabilen Glukosespiegel selbst unter verlängerten Fastenbedingungen aufrechterhalten.
Diese Funktion ist von großer Bedeutung, da das Gehirn fast ausschließlich von Blutzucker genährt wird, sodass schwere Hypoglykämie (sehr niedrige Blutzuckerkonzentrationen) Wissensverlust verursachen können.
Verwandte Krankheiten
Glykogenbezogene Krankheiten erhalten den generischen Namen "Glykogenspeicherkrankheiten".
Diese Krankheiten bilden eine Gruppe erblicher Pathologien, die durch die Ablagerung in den Geweben abnormaler Mengen oder Glykogen -Arten gekennzeichnet sind.
Die meisten Glykogenspeichererkrankungen werden durch ein genetisches Naturdefizit eines der am Glykogenstoffwechsel beteiligten Enzyme verursacht.
Sie werden in acht Typen eingeteilt, von denen die meisten ihre eigenen Namen haben, und jeder von ihnen wird durch ein anderes enzymatisches Defizit erzeugt. Einige sind in sehr frühen Lebensphasen sterblich, während andere während des Trainings von Muskelschwäche und Defizit begleitet werden.
Herausragende Beispiele
Einige der bekanntesten glykogenbezogenen Krankheiten sind die folgenden:
- Von Gierke-Krankheit oder Glykogenspeichererkrankung vom Typ I wird durch ein 6-Phosphatase-Glukosedefizit in Leber und Niere erzeugt.
Es ist durch abnormales Leberwachstum (Hepatomegalie) aufgrund der übertriebenen Akkumulation von Glykogen und Hypoglykämie gekennzeichnet, da die Leber nicht in der Lage ist, Glukose für den Kreislauf zu liefern. Patienten mit dieser Erkrankung haben Wachstumsveränderungen.
- Die Pompe- oder Typ-II-Krankheit ist auf ein α-Defizit (1 → 4) -Glucano 6-Glycosyltransferas in Leber-, Herz- und Skelettmuskeln zurückzuführen. Diese Krankheit ist wie Andersen oder Typ IV vor den zwei Lebensjahren tödlich.
- Mcardle oder Typ V -Krankheit hat ein Muskelphosphorylase -Defizit und wird von Muskelschwäche, verringerter Bewegungstoleranz, abnormaler Akkumulation von Muskelglykogen und Fehlen von Laktat während des Trainings während des Trainings begleitet.
Verweise
- Bhattacharya, k. (2015). Untersuchung und Verwaltung der Leberglykogenspeicherkrankheiten. Translationale Pädiatrie, 4(3), 240-248.
- Dagli, a., Sentner, c., & Weinstein, D. (2016). Glykogenspeicherkrankheit Typ III. Genbewertungen, 1-16.
- Guyton, a., & Hall, j. (2006). Lehrbuch der medizinischen Physiologie (11. Ausgabe.). Elsevier Inc.
- Mathews, c., Van Holde, K., & Ahern, k. (2000). Biochemie (3. Aufl.). San Francisco, Kalifornien: Pearson.
- McKiernan, p. (2017). Pathobiologie des Leberglykogenspeicherwunsches. Curr Pathobiol Rep.
- Murray, r., Bender, d., Botham, k., Kennelly, p., Rodwell, v., & Weil, p. (2009). Harpers illustrierte Biochemie (28. ed.). McGraw-Hill Medical.
- Nelson, d. L., & Cox, m. M. (2009). Lehninger Prinzipien der Biochemie. Omega -Ausgaben (5. Aufl.).
- Rawn, j. D. (1998). Biochemie. Burlington, Massachusetts: Neil Patterson Publishers.
- Tarnopolsky, m. ZU. (2018). Myopathien im Zusammenhang mit Glykogenstoffwechselstörungen. Neurotherapeutika.
- « Argongeschichte, Struktur, Eigenschaften, verwendet
- Bijektive Funktion Was ist, wie ist es getan, Beispiele, Übungen »