

Prionen
- 674
- 116
- Ibrahim Steuk
Prionen sind schlecht gefaltete Proteine, die diese fehlerhaften Informationen an andere Proteine übertragen und übertragbare spongiforme Enzephalopathien verursachen. Quelle: Wikimedia Commons Was sind Prionen?
Der Prionen Sie sind Proteine ohne Genom oder Nukleinsäuren, die als Infektionsmittel wirken. Sie kommen in der normalen Zellmembran vor, nur als schlecht gefaltete Proteine und/oder mit abnormaler dreidimensionaler Struktur.
Diese Proteine sind für mehrere degenerative Erkrankungen und eine sehr hohe Mortalität verantwortlich, die Nervengewebe und Gehirnstruktur beeinflussen.
Sie werden auch als Prionkrankheiten bezeichnet. Zu den wichtigsten, die Menschen betreffen, gehören Kuru, Gerstmann-Sträusler-Scheinker-Krankheit, Creutzfeldt-Jakob-Syndrom und tödliche Familien Schlaflosigkeit.
Prioneigenschaften
- Prons sind Proteinstrukturen, die in Zellmembranen vorhanden sind. Diese Proteine haben eine veränderte Form oder Konformation [PRP (SC)].
- In Bezug auf die Multiplikation wird es durch Umwandlung von Formen erreicht, wie bei der zitternden Krankheit. Bei dieser Krankheit rekrutieren Prionen PRP (C) (prionische Proteine unbeaufsichtigter Konformation), um die Umwandlung in die PRP (SC) -Isoform zu stimulieren.
- Diese ungewöhnlichen Proteine, die sich ausbreiten können, haben keine Nukleinsäuren. Der Beweis dafür ist, dass sie gegen X -Strahlen und ultraviolette Strahlung resistent sind. Diese Mittel brechen leicht Nukleinsäuren ab.
- Prionische Proteine, von denen Prionen (PRP) zusammengesetzt sind, finden sich nicht nur von Menschen, sondern auch von anderen gesunden Wirbeltieren im Körper.
- Einige Forscher haben es geschafft zu demonstrieren, dass diese Proteine bei Mäusen die myelinische Reparatur in Zellen des peripheren Nervensystems aktivieren. Es wurde auch gezeigt, dass das Fehlen dieser die Demyelinisierung solcher Nervenzellen verursacht.
Prionstruktur
Das Wissen über die Struktur der Prionen liegt hauptsächlich in den Untersuchungen, die in den Bakterien durchgeführt werden Escherichia coli.
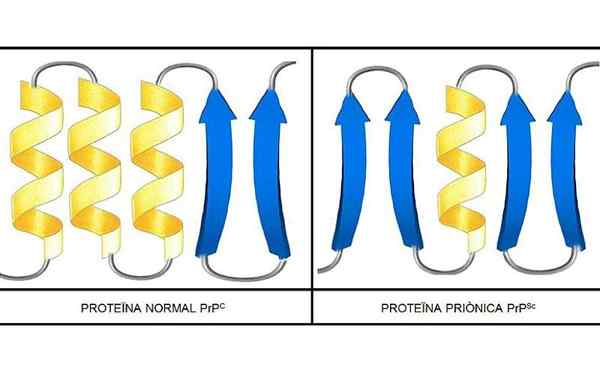
Die Studien wurden durchgeführt, dass die Polypeptide in Ketten -PRP (c) (normal) und PRP (SC) (infektiös) in der Zusammensetzung von Aminosäuren identisch sind, sich jedoch in der 3D -Konformation und in der Faltung dieser unterscheiden.
PRP (c)
Diese nicht infektiösen Prionen vorhanden, beim Menschen, 209 Aminosäuren. Sie haben eine Disulfidverbindung. Seine Struktur ist alpha-helikoidal, was bedeutet, dass es spiralförmige Aminosäuren (Alpha-Propeller) und nur wenige flache Aminosäurestränge (Beta-Blätter) hat.
Es kann Ihnen dienen: Hilfswissenschaften der BiologieDieses Protein kann nicht durch Zentrifugation getrennt werden, was bedeutet, dass es nicht sedimentierbar ist. Es kann leicht durch die breite Protease des breiten Spektrums als Proteinase K verdaut werden.
PRP (SC)
Es ist ein infektiöses Protein, das PRP (C) in infektiöse PRP (SC) -Isoformen verwandelt.
Über seine 3D -Struktur ist jedoch sehr wenig bekannt. Es ist jedoch bekannt, dass es nur wenige helikale Formen und mehr flache Stränge oder Beta -Blätter hat. Die Veränderung in Richtung Isoform ist das, was als grundlegendes Ereignis von Prionerkrankungen bekannt ist.
Prionfunktionen
Zellprion -Proteine [PRP (C)] befinden sich auf der Zelloberfläche einer Vielzahl von Organen und Geweben. Über die physiologischen Funktionen von Prionen im Körper ist sehr wenig bekannt.
Trotzdem geben Erfahrungen in Mäusen mögliche Funktionen an, wie z. B.:
Mit metabotropen Glutamatrezeptoren
Es wurde gezeigt, dass PRP (C) mit Glutamatrezeptoren (Ionotropika und Metabotropika) wirken. Der PRP (c) nimmt als Rezeptor der synaptotoxischen Oligomere des Zellpeptids der Zelloberfläche teil.
In der embryonalen Entwicklung
In Murinae -Familienmäusen wurde festgestellt.
Dies zeigt an, dass sie während der Entwicklung dieser kleinen Säugetiere eine Rolle spielen. Papier, das nach Angaben der Forscher mit der Regulierung der Neuritogenese (Produktion von Axonen und Dendriten von Neuronen) zusammenhängt, bezieht sich auf.
Sie wirken auch im axonalen Wachstum. Diese Prionproteine sind sogar an der Entwicklung des Kleinhirnkreislaufs beteiligt. Aus diesem Grund wird angenommen, dass das Fehlen dieser Prionen PRP (c) eine Verzögerung bei der motorischen Entwicklung von Nagetieren beinhaltet.
Neuroprotektor
In Studien zur Überexpression von PRP (c) aufgrund der Ausrichtung von Genen wurde festgestellt, dass das Fehlen dieser Prionen an einigen Orten im Gehirn Probleme mit der Blutbewässerung verursacht (akute Gehirnischämie).
Dies bedeutet, dass Prionproteine als Neuroprotektoren fungieren. Darüber hinaus wurde gezeigt, dass die Überexpression von PRP (C) Läsionen durch Ischämie verringern oder verbessern kann.
Periphäres Nervensystem
Vor kurzem wurde die physiologische Funktion von PRP (C) bei der Aufrechterhaltung des peripheren Myelins entdeckt.
Kann Ihnen dienen: Dystrophin: Eigenschaften, Struktur und FunktionenWährend einer Laborstudie wurde festgestellt, dass in Abwesenheit von Prionprotein Labormäuse Nervenmangel entwickelten.
Zelltod
Es gibt einige Prionproteine und befinden sich in anderen Körperteilen als im Gehirn.
Die Funktionen solcher Proteine sind es, den Zelltod zu initiieren, zu regulieren und/oder zu kontrollieren, wenn der Körper angegriffen wird (z. B. durch Vulonen), wodurch die Ausbreitung des Erregers verhindert wird.
Diese besondere Funktion dieser Proteine lässt die Forscher über die mögliche Bedeutung von nicht infektiösen Prionen im Kampf gegen Krankheitserreger nachdenken.
Langzeitgedächtnis
Eine Studie, die am Stowers Institute in Missouri, EE, durchgeführt wurde. UU., Er zeigte, dass PRP -Prionen eine Funktion bei der Aufrechterhaltung des Langzeitgedächtnisses haben können.
Die Studie ergab, dass bestimmte Prionproteine kontrolliert werden können, um an der Aufrechterhaltung der physiologischen Funktionen des Langzeitgedächtnisses zu arbeiten.
Mutterzellerneuerung
Eine Untersuchung zu Prionproteinen, die in Stammgewebezellen exprimiert werden, ergab, dass alle diese Stammzellen (hämatopoetisch) Prionproteine in ihrer Zellmembran exprimieren. Es wird also angenommen, dass sie am komplexen und sehr wichtigen Prozess der Zellerneuerung teilnehmen.
Durch Prionen verursachte Krankheiten
Die häufigsten Prionerkrankungen sind:
Creutzfeldt-Jakob (EUJ) -Rehne (EuGH)
Es gilt als die häufigste Prionerkrankung beim Menschen und ist eine kosmopolitische Pathologie, dh weltweite Verteilung. Es kann Erbschaft (Familie), sporadisch oder ansteckend auftreten.
Gerstmann-Sträusler-Scheinker-Krankheit
Es handelt sich um eine Krankheit, die von Prionen in einem ererbten ansteckenden oder autosomal dominanten enzephalen Prozess verursacht wird. Die Krankheit manifestiert sich bei Menschen von 40 bis 60 Jahren.
Prionopathie mit variabler Proteaseempfindlichkeit
Es ist eine sehr seltene Krankheit, bis zu dem Punkt, dass sein Auftreten von 2 bis 3 Fällen pro 100 Millionen Einwohner beträgt. Pathologie ist ähnlich wie die Gerstmann-Stränussler-Scheinker-Krankheit.
Tödliche Schlaflosigkeit
Es ist eine erbliche oder vertraute Krankheit, obwohl sie auch sporadisch auftreten kann. Es ist bekannt, dass die Krankheit auf eine dominante erbliche oder autosomale Mutation zurückzuführen ist.
Kann Ihnen dienen: Endemische ArtenKuru
Diese Prionerkrankung wurde nur bei Bewohnern von Papua -Neuguinea nachgewiesen. Es ist eine Krankheit im Zusammenhang mit Kannibalismus und der kulturellen Tradition des Duellritus für die Toten, wo diese Menschen das menschliche Gehirn essen.
Krankheiten bei Tieren
Unter den von Prion bei Tieren produzierten Pathologien befindet sich eine spongiforme Enzephalopathie -Rinder. Diese Krankheit hat in Europa, in der öffentlichen Gesundheit, in der von Tieren und in der Wirtschaft der betroffenen Länder verwüsten.
Andere Krankheiten bei Tieren sind Scrapy, übertragbare Enzephalopathie des Nerzes, chronische Verschleißerkrankung (bei Hirsch) und katzenpolige Spongiform -Enzephalopathie.
Diese Krankheiten haben, wie die beim Menschen vorgestellten Krankheiten, eine wirksame Behandlung. Daher ist die Prävention von grundlegend.
Behandlungen
Bisher ist kein Heilmittel für Prionkrankheiten bekannt. Die Behandlung ist symptomatisch. Den Patienten wird empfohlen, die Palliativversorgung zu planen, und genetische Analysen und Ratschläge für Familienmitglieder werden empfohlen.
Eine Vielzahl von Medikamenten wurde bei Patienten mit Prionerkrankungen wie antiviralem Antitumor getestet.
Derzeit gibt es jedoch keine Hinweise darauf.
Verhütung
Die Prons sind gegen eine Vielzahl von physikalischen und chemischen Veränderungen resistent. Es werden jedoch verschiedene Techniken verwendet, um die Verschmutzung von Patienten mit kontaminierten chirurgischen Instrumenten zu vermeiden.
Zu den am häufigsten verwendeten Techniken gehört es, Geräte in einem Autoklav bei 132 ° C für eine Stunde zu sterilisieren und die Instrumente dann mindestens eine Stunde in Natriumhydroxid zu übertreffen.
Andererseits hat die Weltgesundheitsorganisation (WHO) Maßnahmen entwickelt, um die Ausbreitung von Prionkrankheiten zu vermeiden. Diese Organisation legt Regeln für das Management verbotener oder potenziell riskantes Gewebe wie: Augen, Gehirn, Darm, Mandeln und Rückenmark fest.
Verweise
- Prion, Infektionsmittel. Von Britannica geborgen.com.
- Was ist ein Prion? Von Scientifican geborgen.com.
- Prion. Abgerufen von.Wikipedia.Org