

Succinato -Dehydrogenasestruktur, Funktion, Regulation, Krankheiten
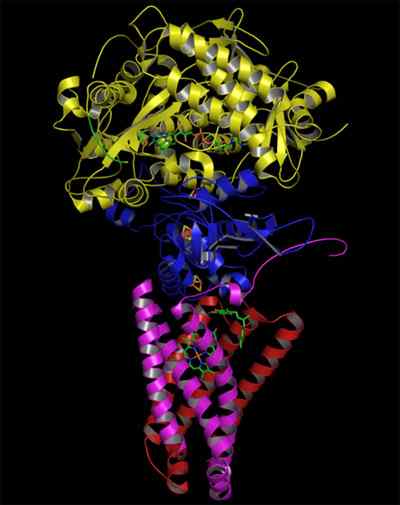
- 3110
- 183
- Frederike Birkemeyer
SUccinato -Dehydrogenase (SDH), auch als Komplex II der Elektronentransportkette bekannt, ist ein mitochondrialer Proteinkomplex mit enzymatischer Aktivität, der sowohl im Krebszyklus als auch in der Elektronenförderkette (Zellatmung) funktioniert.
Es ist ein Enzym, das in allen aeroben Zellen vorhanden ist. In Eukaryoten ist es ein Komplex.
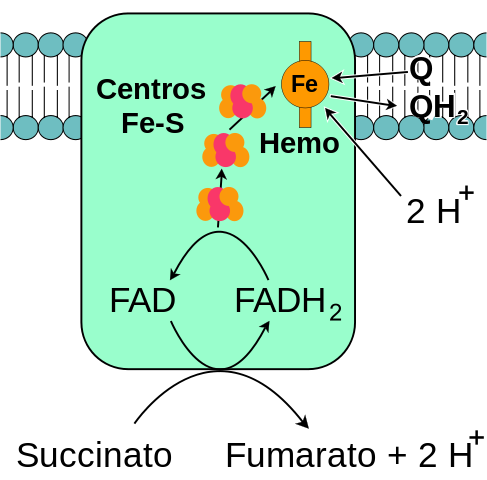 Allgemeines Schema der mitochondrialen Komplex -Succinat -Dehydrogenase (Quelle: Mich, basierend auf der Fvasconcellos -Vektorisierung. / Public Domain, über Wikimedia Commons)
Allgemeines Schema der mitochondrialen Komplex -Succinat -Dehydrogenase (Quelle: Mich, basierend auf der Fvasconcellos -Vektorisierung. / Public Domain, über Wikimedia Commons) Die um 1910 entdeckte komplexe Succinat -Dehydrogenase, die 1954 von Sänger und Kearney zum ersten Mal gereinigt wurde, wurde aus mehreren Gründen ausführlich untersucht:
- Es funktioniert sowohl im Krebszyklus (Zitronensäurzyklus als auch Tricarbonsäurezyklus) und in der Elektronentransportkette (katalysiert die Oxidation von Succinat zu Fumarat)
- Seine Aktivität wird durch verschiedene Aktivatoren und Inhibitoren reguliert und
- Es ist ein Komplex, das mit: Eisen verbunden ist, nicht mit einer Hämogruppe, Labylsulfur und Dyukleotiden von Flavina Adenina (FAD) verbunden
Es wird vom Kerngenom codiert und es wurde nachgewiesen, dass Mutationen in den vier Genen, die jede seiner Untereinheiten (A, B, C und D) kodifizieren der Sicht der physischen Integrität der Menschen.
[TOC]
Struktur
Das Enzymkomplex -Succinat -Dehydrogenase wird durch vier Untereinheiten (Heterotarámero) gebildet, die vom Kerngenom kodiert werden. Daher ist es der einzige Komplex der oxidativen Phosphorylierung in der Elektronenförderkette.
Darüber hinaus ist dieser Komplex der einzige, der Protonen nicht durch die interne mitochondriale Membran während ihrer katalytischen Wirkung pumpt.
Laut Studien, die auf dem enzymatischen Komplex von Schweinherzzellen basieren, besteht die komplexe Succinat -Dehydrogenase aus:
- A "Kopf" Hydrophil die sich von der internen mitochondrialen Membran bis zur mitochondrialen Matrix und
- A "Linie" Hydrophob die in die innere mitochondriale Membran eingebettet ist und ein kleines Segment hat, das auf den löslichen intermembranischen Raum der Mitochondrien projiziert wird
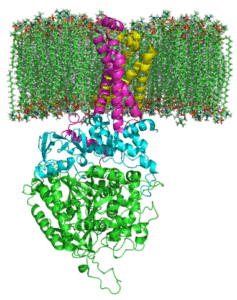 Struktur der komplexen Succinat-Dehydrogenase (Quelle: Zephyris bei der englischen Sprache Wikipedia/CC BY-SA (http: // CreePecommons.Org/lizenzen/by-sa/3.0/) über Wikimedia Commons)
Struktur der komplexen Succinat-Dehydrogenase (Quelle: Zephyris bei der englischen Sprache Wikipedia/CC BY-SA (http: // CreePecommons.Org/lizenzen/by-sa/3.0/) über Wikimedia Commons) Hydrophile Portionsstruktur
Der hydrophile Kopf besteht aus SDHA (70 kDa) und SDHB (27 kDa) (SDH1 und SDH2 in Hefen), und dies umfasst das katalytische Zentrum des Komplexes.
SDHA- und SDHB -Untereinheiten enthalten Redox -Cofaktoren, die an der Übertragung von Elektronen auf das Ubiquinon beteiligt sind (Coenzym Q10, ein Molekül, das Elektronen zwischen den Atemkomplexen I, II und III transportiert).
Die SDHA-Untereinheit hat einen FAD-Cofaktor (ein Coenzym, das an Oxid-Reduktionsreaktionen teilnimmt) zusammen mit seiner Struktur, genau an der Stelle der Verbindung für Succinat (das Hauptsubstrat des Enzyms).
Die SDHB-Untereinheit verfügt. Einer der Zentren, 2FE-2S, befindet sich in der Nähe der Modeerscheinung der SDHA-Untereinheit und die anderen (4Fe-4s und 3Fe-4s) befinden sich an die erste.
Kann Ihnen dienen: PhylogenieEs ist zu beachten, dass strukturelle Studien darauf hinweisen.
Hydrophobe Portionsstruktur
Die membranale Domäne des Komplexes besteht, wie angegeben, aus SDHC (15 kDA) und SDHD (12-13 kDA) (SDH3 und SDH4 in Hefen), die umfassende Membranproteine sind, die jeweils 3 transmembranistische Membranproteine sind.
Diese Domäne enthält einen HEMO -Teil B Verbunden Sie in der Grenzfläche zwischen den SDHC- und SDHD -Untereinheiten, wo jede der beiden Histidinliganden, die sie zusammenhalten.
In diesem Enzym wurden zwei Gewerkschaftsstandorte für Ubiquinona festgestellt: eine der großen Affinität und eine der niedrigen Affinität.
Die hochaffinitätsberechtigte Stelle, bekannt als QP (P by proximal) Es ist Gesicht zur Mitochondrienmatrix und wird durch bestimmte Aminosäurereste gebildet, die sich in den Untereinheiten SDHB, SDHC und SDHD befinden.
Die Site mit niedriger Affinität, auch genannt Qd (D von distal) Es befindet sich im Teil der inneren mitochondrialen Membran, in der der Komplex näher am Zwischenraum des Intermembrans eingefügt wird, dh weiter von der Organelle Matrix entfernt.
Zusammen hat der Gesamtkomplex ein Molekulargewicht von fast 200 kDa und es wurde festgestellt, dass er ein Verhältnis von 4 hat.2-5.0 Flavin-Nanomole pro Milligramm Protein und 2-4 g Eisen für jeden Mol Flavina.
Funktion
Der enzymatische Succinat -Komplex -Dehydrogenase erfüllt eine wichtige Funktion in den Mitochondrien, da nicht nur am Krebszyklus beteiligt ist (wo sie am Abbau von Acetyl -CoA beteiligt ist), sondern auch Teil der Atemwegskette, grundlegend für die Energieproduktion ATP -Shaped.
Mit anderen Worten, es ist ein Schlüsselenzym für den Vermittlungsstoffwechsel und die Aerobic -Produktion von ATP.
- Es ist verantwortlich für die Oxidation des Succinat zu Fumarat im Zitronensäurzyklus
- Es füttert den Komplex III der Elektronenförderkette mit den Elektronen, die aus der Oxidation des Succinat abgeleitet sind, was dazu beiträgt, Sauerstoff zu reduzieren und Wasser zu bilden
- Der Elektronentransport erzeugt einen elektrochemischen Gradienten durch die innere mitochondriale Membran, die die ATP -Synthese begünstigt
Alternative können Elektronen verwendet werden, um Moleküle aus einem „Pool“ von Ubiquinonas zu reduzieren und die notwendigen Reduzierer zu erzeugen, um Superoxidanionen zu reduzieren, die aus derselben Atemkette stammen oder aus exogenen Quellen stammen.
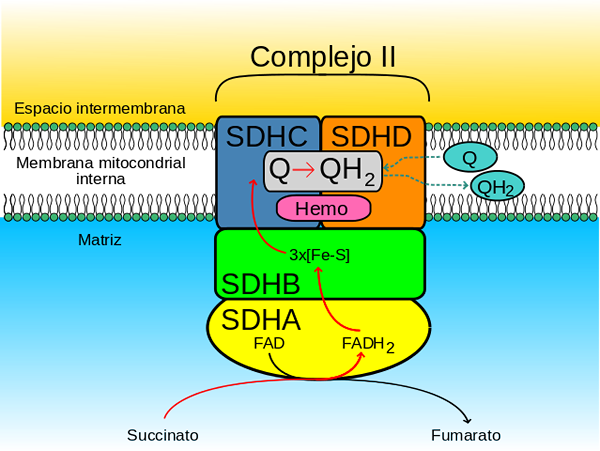 Succinate Complex Degidrogenasa (Quelle: JohnHFST / Public Domain, über Wikimedia Commons)
Succinate Complex Degidrogenasa (Quelle: JohnHFST / Public Domain, über Wikimedia Commons) Wie handelt es sich?
Die Untereinheit A des Komplexes (der kovalent mit dem Fad Coenzym verbunden ist) verbindet die Substrate, Fumarat und Succinat sowie seine physiologischen Regulatoren Oxalacetat (Wettbewerbsinhibitor) und ATP.
Die ATP verdrängt die Vereinigung zwischen Oxalacetat und dem SDH -Komplex und dann die Elektronen, die vom Succinat bis zur SDHA -Untereinheit "vorbei sind".
Kann Ihnen dienen: Myosin: Eigenschaften, Struktur, Typen und FunktionenAus der Untereinheit B erreichen diese Elektronen die Hämostellen B der SDHC- und SDHD.
Der elektronische Fluss vom Succinat durch diese Transporter und sogar der endgültige Akzeptor, der Sauerstoff ist, ist mit der Synthese von 1 gekoppelt.5 ATP -Moleküle für jedes elektronische Drehmoment durch Atemkettenphosphorylierung.
Inzima -Defekte
Es wurde berichtet, dass Mutationen im Gen, die für die Untereinheit A der komplexen Succinat -Dehydrogenase codiert, während der Kindheit Enzephalopathien verursachen können.
Verordnung
Die Aktivität der komplexen Succinat -Dehydrogenase kann durch post -translationale Modifikationen wie die reguliert werden Phosphorylierung und das Acetylierung, Obwohl auch eine aktive Zentrumshemmung auftreten kann.
Die Acetylierung einiger Lysinabfälle kann die Aktivität dieses Enzyms verringern und dieses Verfahren wird durch ein als SIRT3 bekanntes Acetylase -Enzym durchgeführt. Die Phosphorylierung hat den gleichen Effekt auf das Enzym.
Zusätzlich zu diesen Modifikationen wird der SDH -Komplex auch durch Vermittler des Krebszyklus reguliert, insbesondere durch die Oxalacetat und das Succinat. Oxalacetat ist ein starker Inhibitor, während Succinat die Dissoziation von Oxalacetat bevorzugt und als Aktivator fungiert.
Impulsmangel Dehydrogenase
Die Dehydrogenase der Succinatmangel ist eine Anomalie- oder mitochondriale Atemkettenstörung. Dieser Mangel wird durch SDHA (oder SDHAF1), SDHB, SDHC und SDHD -Mutationen verursacht.
Unterschiedliche Untersuchungen haben homozygote und heterozygote Mutationen in diesen Genen, insbesondere SDHA, gezeigt. Die Mutationen dieser Gene verursachen Aminosäuresubstitutionen im Protein (in einem der SDHA, B, C oder D) oder fehlschlagen, die ungewöhnlich kurze Proteine kodifizieren.
Daher führen Aminosäure -Substitutionen und ungewöhnlich kurze Proteincodierung zu Störungen oder Veränderungen des SDH -Enzyms, die zu einem Versagen der optimalen Kapazität von Mitochondrien führen. Dies nennen Wissenschaftler als mitochondriale Atemwegsstörung.
Diese Störung kann in vielerlei Hinsicht im Menschen phänotypisch ausgedrückt werden. Am bekanntesten sind: Mangel oder mangelnde sprachliche Entwicklung, spastischer Quadruplex, unfreiwillige Kontraktionen (Dystonie), Muskelschwäche und Myokardiopathie unter anderem verwandte Probleme.
Einige Patienten mit Dehydrogenase mit Succinatmangel können die Krankheit von Leight oder das Kearns-Saire-Syndrom manifestieren.
Wie wird dehydrogenierter Succinatmangel erkannt??
Bestimmte Studien deuten auf die Verwendung qualitativer Tests und Analysen sowie quantitative, enzymatische biochemische Analyse der respiratorischen Kette vor. Andere deuten andererseits die vollständige Ausdehnung durch die Polymerasekettenreaktion (PCR) der Exons der untersuchten Untereinheiten und dann die jeweilige Sequenzierung vor.
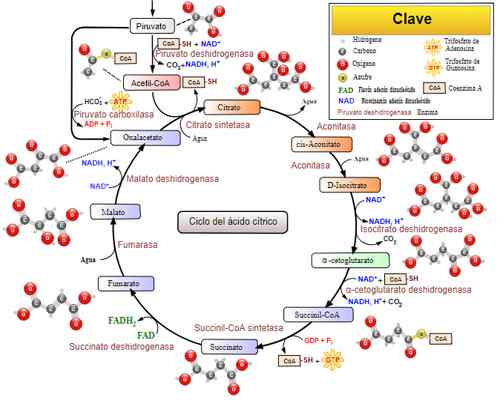 Tricarbonsäurezyklus (Krebszyklus). Genommen und bearbeitet von: Narayanese, Wikiuserpedia, Yassinemrabet, Totobaggins (übersetzt in Spanisch von Alejandro Porto) [CC BY-SA 3.0 (https: // creativecommons.Org/lizenzen/by-sa/3.0)]].
Tricarbonsäurezyklus (Krebszyklus). Genommen und bearbeitet von: Narayanese, Wikiuserpedia, Yassinemrabet, Totobaggins (übersetzt in Spanisch von Alejandro Porto) [CC BY-SA 3.0 (https: // creativecommons.Org/lizenzen/by-sa/3.0)]]. Verwandte Krankheiten
Es gibt viele phänotypische Ausdrücke, die durch mitochondriale Atemkettenstörungen aufgrund von Dehydrogenase von Succinatmangel erzeugt werden. Wenn es jedoch um Syndrome oder Krankheiten geht.
Kann Ihnen dienen: Die 8 wichtigsten biogeochemischen Zyklen (Beschreibung)Leight -Syndrom
Es ist eine fortschreitende neurologische Erkrankung, die mit Mutationen im Kerngenom (in diesem Fall von Dehydrogenase-Succinat) verbunden ist, die den Pyruvat-Dishydrogenase-Komplex zum oxidativen Phosphorylierungsweg beeinflussen.
Die Symptome treten vor dem ersten Jahr des Einzelnen auf, aber in atypischen Fällen wurden die ersten Symptome während der Adoleszenz beobachtet.
Zu den am häufigsten beobachteten Symptomen gehören: Hypotonie mit Verlust der kephalischen Kontrolle, unfreiwilligen Bewegungen, rezidivierendes Erbrechen, Atemproblemen, Unfähigkeit, unter anderem unter anderem die Augenblutzellen, Pyramiden und Extrapiramiden unter anderem zu bewegen. Anfälle sind nicht sehr häufig.
Es ist möglich, dass die Krankheit in vorgeburtlichen Diagnosen festgestellt werden kann. Eine spezifische Heilung oder Behandlung ist nicht bekannt, aber einige Spezialisten schlagen Behandlungen mit bestimmten Vitamin oder Cofaktoren vor.
Magen -Darm -Stroma -Tumor (GIST)
Allgemein als GIST bezeichnet, handelt. Es wird angenommen, dass die Ursache dieser.
Andere Überlegungen zur Ursache von GIST sind die Mutationen bestimmter Arten von Genen, die nach Ansicht einiger Autoren 90% der Tumoren verursachen. Die beteiligten Gene sind: Kit, PDGFRA -Gene, Dehydrogenase -Succinat (SDH) - schlecht.
Die Succinat -Dehydrogenase (SDH) - schlecht, tritt hauptsächlich bei jungen Frauen auf, produziert Magentumoren und erzeugt mit relativer Frequenz Metastasen in Lymphknoten. Bei Kindern tritt ein kleiner Prozentsatz auf.
Kearns-Sayre-Syndrom
Es wurde festgestellt. Diese Krankheit hängt mit mitochondrialen Störungen zusammen und ist durch das Fehlen einer Bewegung von Augenballons gekennzeichnet.
Andere Merkmale dieser Krankheit sind Pigments -Retinitis, Taubheit, Kardiomyopathie und Bedingungen des Zentralnervensystems. Normalerweise werden diese Symptome beobachtet, bevor der Patient 20 Jahre alt wird. Es ist keine pränatale Diagnose für diesen Zustand bekannt.
Auch ist für diese Krankheit auch heilt. Die Behandlung ist palliativ, dh sie arbeitet nur daran, die Auswirkungen der Krankheit zu verringern, nicht die Heilung. Andererseits ist die Lebenserwartung relativ normal, obwohl es von der Anzahl der betroffenen Organe und der erhaltenen medizinischen Versorgung abhängt, relativ normal ist.
Verweise
- Ackrell, geb. ZU., Kearney, e. B., & Sänger, t. P. (1978). [47] Säugetier -Succinat -Dehydrogenase. In Methoden in der Enzymologie (vol. 53, pp. 466-483). Akademische Presse.
- Brère, j. J., Favier, J., Ghouzzi, v. UND., Djoudi, f., Benit, p., Gimenez, a. P., & Rustin, p. (2005). Succinat -Dehydrogenase -Deficizität beim Menschen. Zell- und molekulare Biowissenschaften CMLS, 62 (19-20), 2317-2324.
- Cecchini, g., Schröder, ich., Gunalus, r. P., & Maklashina und. (2002). Succinat -Dehydrogenase und Fumatre -Redaktase aus Escherichia coli. Biochimica et Biophysica Acta (BBA) -Bioenergetics, 1553 (1-2), 140-157.
- Hassefi und., & Davis, k. ZU. (1971). Succinat -Dehydrogenase. Yo. Reinigung, molekulare Eigenschaften und Unterstruktur. Biochemie, 10 (13), 2509-2516.
- Hederstedt, l. ZU. R. S., & Rutberg, l. ZU. R. S. (1981). Succinat-Dehydrogenase-A vergleichende Überprüfung. Mikrobiologische Bewertungen, 45 (4), 542.
- Nelson, d. L., Lehninger, a. L., & Cox, m. M. (2008). Lehninger Prinzipien der Biochemie. Macmillan.
- Rutter, j., Winge, d. R., & Schiffman, J. D. (2010). Succinat-Dehydrogenase-Asembly, Regulation und Rolle bei menschlicher Diew. Mithochondrion, 10 (4), 393-401.
- « Essentielle Fettsäuren Funktionen, Wichtigkeit, Nomenklatur, Beispiele
- Regeln -T -Funktionen, damit es dient, Beispiele »